Documentation
The PUResNetV2.0 webserver is a powerful tool designed to offer comprehensive predictive and visualization capabilities for protein binding pocket analysis. The platform is built on Django, ensuring a user-friendly interface, and streamlined operations for molecular biology researchers.
Webserver Implementation
The webserver operates in three main stages:
- Predictive Options: Users can upload a Protein Data Bank (PDB) file, enter a PDB ID, or a UniProt ID. The prediction mode can be set to treat the input as a single entity, each chain as a separate entity, or comma-separated chains as a single entity.
- Backend Processing: The webserver processes the PDB file, creating a sparse tensor. It uses PUResNetV2.0 to predict binding pockets, then post-processes these predictions for visualization.
- Results Visualization: The predicted protein structure and its binding pockets are displayed using JSmol. The amino acids in each pocket are identified and can be downloaded as a PDB file.
User Input Options
Users can input protein structure data in three ways:
- Upload a Protein Structure in .pdb File Format: Users can directly upload a protein structure in the widely accepted PDB file format. This method is most suitable for custom-designed models or structures not yet available in public databases.
- Provide a Valid PDB ID: Inputting a valid PDB ID allows the tool to fetch the associated protein structure from the RCSB Protein Data Bank. This method is ideal for users wishing to analyze publicly available protein structures.
- Provide a Valid UniProt ID: By providing a valid UniProt ID, the tool can retrieve the protein structure from the AlphaFold database. This option is useful when the protein's experimental structure is unavailable, but a predicted structure from AlphaFold exists.
Prediction Mode
Users have three options for binding site prediction modes:
- Each Chain as Individual Entity: In this mode, each chain in the protein structure is treated separately, and binding site prediction is performed independently for each chain.
- Whole Structure as Single Entity: The entire protein structure, including all chains, is treated as a single entity for binding site prediction.
- Chain (Comma Separated Values) or Single Entity: Users can specify which chains to include in the binding site prediction by providing a comma-separated list of chain identifiers. These specified chains are treated as a single entity during the prediction process.
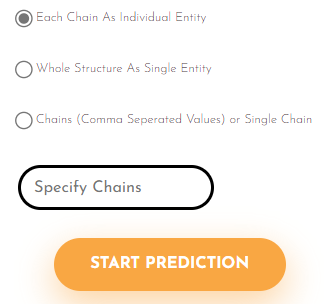
Visualization and Results Access
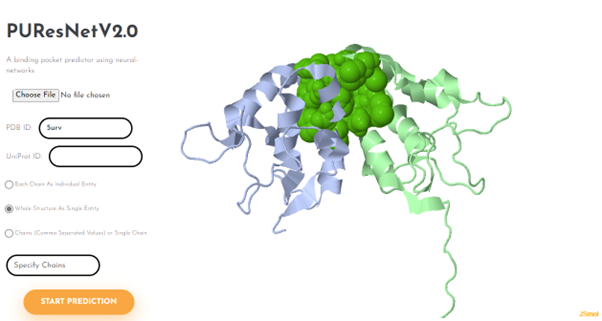
After prediction, the protein structure and predicted binding pockets are visualized using JSmol, an open-source HTML5-based molecular viewer.

Each job performed by the user is assigned a unique identifier. Using unique identifier user can access their past results from https://nsclbio.jbnu.ac.kr/puresnet/*identifier. Replace *identifier with your identifier
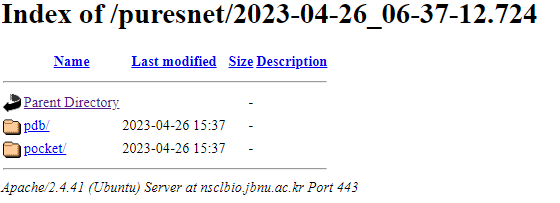
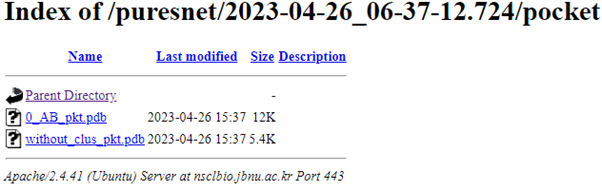
Users can access their predictions by clicking on this identifier, which opens a directory containing individual predicted pockets and a PDB file (without_clus_pkt.pdb) with all predictions without clustering.

For each predicted pocket, detailed residue information is available.
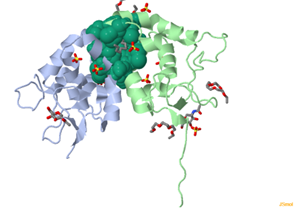
The "HET ATOM" button reveals any non-standard atoms or molecules in the structure.
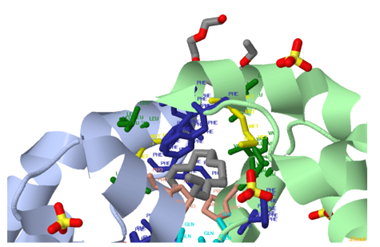
Each amino acid has a corresponding button, and when clicked, the respective residues are displayed in the structure.

The "Pocket checkbox" allows selective display or hiding of predicted pockets, useful for focusing on specific regions.
Users can download individual pockets by clicking on the respective “*_pkt” button.
Code Availability
The complete code for this project is publicly accessible and can be found at https://github.com/jivankandel/PUResNetV2.0.git It comes with comprehensive documentation for ease of understanding and utilization.
Bibliography
- Hanson, R. M., Prilusky, J., Renjian, Z., Nakane, T. & Sussman, J. L.
- Jumper, J. et al., Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
- Varadi, M. et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research 30, D439–D444 (2021).